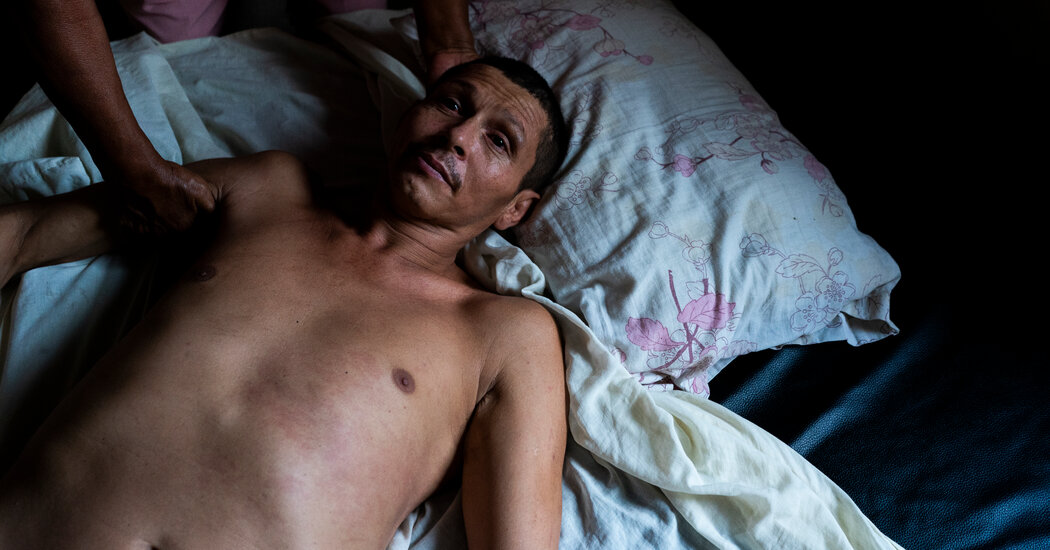
José Echeverría pasa días de inquietud en una silla de metal reforzada con tablas y acolchada con un trozo de espuma que su madre, Nohora Vásquez, ajusta constantemente para que esté cómodo. La silla se está aflojando y pronto se caerá a pedazos. La enfermedad de Huntington, que ocasiona que José mueva la cabeza y las extremidades sin control, ya ha dejado destrozado el armazón de una cama. A sus 42 años, sigue estando fuerte.
Nohora Esther Echeverría, de 37 años y hermana de José, vive con su madre y su hermano. Con solo dos años de enfermedad, sus síntomas son más leves que los de él, pero tiene miedo de caminar por las calles empinadas de su ciudad pues sabe que podría caerse. Un cartel en la puerta principal anuncia la venta de un ron que no existe. Los escasos recursos de la familia ahora se destinan a la comida —José y Nohora Esther deben comer con frecuencia o perderán peso rápidamente— y a suministros médicos, como una costosa crema para la piel de José.
La enfermedad de Huntington es una afección neurodegenerativa hereditaria causada por un exceso de repeticiones de tres componentes básicos del ADN —citosina, adenina y guanina— en un gen llamado huntingtina. La mutación da lugar a una versión tóxica de una proteína cerebral clave, y la edad de una persona en el momento de aparición de los síntomas está relacionada, aproximadamente, con el número de repeticiones que porta. Los primeros síntomas pueden incluir alteraciones del estado de ánimo —Vásquez recuerda cómo su difunto marido había echado a los niños de sus camas, obligándola a dormir con ellos en el bosque— y sutiles movimientos involuntarios, como las rotaciones de las delicadas muñecas de Nohora Esther.
La enfermedad es relativamente rara, pero a finales de la década de 1980, un neurólogo colombiano, Jorge Daza, empezó a observar un sorprendente número de casos en la región donde vive Vásquez, un conjunto de ciudades costeras y de montaña cerca de Barranquilla. Al mismo tiempo, científicos estadounidenses dirigidos por Nancy Wexler trabajaban con una familia aún más numerosa con Huntington en la vecina Venezuela, reuniendo y estudiando miles de muestras de tejido para identificar la mutación genética responsable.
Ahora se cree que esta región colombiana alberga la segunda familia extendida más numerosa que padece Huntington. Sus integrantes despiertan un gran interés científico porque albergan pistas sobre los modificadores genéticos y los posibles tratamientos de la enfermedad de Huntington. Sin embargo, desde la prematura muerte de Daza en 2014, han estado aislados de un mundo de prometedores tratamientos experimentales, asesoramiento genético y, a menudo, atención médica básica. Vásquez, como otros de su generación, a veces llama a la enfermedad por su nombre del siglo XVI, el mal de San Vito.
Los miembros de la familia suelen proceder de los segmentos más pobres de esta sociedad costera: personas que se dedican a la pesca, la limpieza de hoteles playeros o la agricultura. Universidades y funcionarios de sanidad se han presentado intermitentemente para tomarles muestras de sangre, pero no se ha publicado ningún estudio epidemiológico, clínico o genético definitivo. Pocos o ningún individuo en riesgo sabe si es portador de la mutación. Los científicos intentan ahora hacer lo correcto por ellos, con la esperanza de que no sea demasiado tarde.
‘Como empezar de cero’
En los últimos años, un grupo de investigadores de la Universidad Simón Bolívar, en Barranquilla, ha asumido la ardua tarea de reavivar los estudios clínicos y genéticos que se estancaron tras la muerte de su colega Daza. Según el neuropsicólogo Johan Acosta, que dirige la iniciativa, ha sido “como empezar de cero”.
En los pueblos y aldeas, los investigadores encontraron a las familias con Huntington recelosas y cansadas. Casi todos afirmaron que en el pasado les habían tomado una muestra de sangre y que no sabían para qué se había usado. Los actos organizados por los investigadores tuvieron escasa asistencia, por lo que tuvieron que “llegar a ciertas familias, llegar a sus casas, tener un contacto más directo”, relató la neuropsicóloga Elsy Mejía.
El proyecto del grupo, patrocinado por el gobierno colombiano, se enfoca en los síntomas no motores y más sutiles de la enfermedad. Los investigadores realizan evaluaciones clínicas e imágenes cerebrales en personas con síntomas iniciales o presintomáticos, así como en no portadores con antecedentes familiares de Huntington. Ya se ha recogido ADN de casi 300 individuos, con el cual los científicos esperan identificar vínculos genéticos con patrones de síntomas tempranos.
Los investigadores han prometido compartir los resultados de sus estudios con los miembros de la familia, así como los resultados de las evaluaciones clínicas, aunque no pueden revelar los resultados de las pruebas genéticas; eso debe esperar hasta que se establezca un asesoramiento. Ofrecen ayuda para conseguir los medicamentos y organizan en el lugar atención clínica de un día de duración en las que cualquier miembro de la familia puede consultar a un médico, por cualquier motivo.
En 2021, el equipo elaboró un manual sobre la enfermedad y su historia en la región, dirigido a los profesionales locales de la salud, que en muchos casos saben poco sobre ella. Esperan que la universidad participe pronto en Enroll-HD, una plataforma mundial para estudiar a personas con enfermedad de Huntington y facilitar ensayos clínicos.
También saben que si ellos —u otros investigadores— dejan de perseverar con los familiares, todo estará perdido. “No es un trabajo solo de ciencias exactas, es un trabajo de ciencias sociales”, afirma Pedro Puentes, neuropsicólogo y líder del grupo de investigación. “Ellos esperan algo” a cambio de lo que han brindado, añadió. “Y si te das cuenta, realmente no se ha hecho nada al respecto”.
Mari y Julieta
Julieta Echeverría, de 23 años, cría a dos niños pequeños en una casa situada sobre un arroyo arenoso al que la gente arroja desperdicios, ya que no hay caminos preparados para un camión recolector de basura. El techo y las paredes de su casa están muy agrietados y cuando llueve hay que desalojar una de las habitaciones.
El año pasado, durante meses, Echeverría y su esposo, un pescador, cuidaron a su tío Nelson Echeverría, soltero, en las últimas fases de la enfermedad de Huntington. Los padres de ella, agricultores, no podían ocuparse de Nelson porque su padre también está enfermo, aunque aún no ha acudido a un neurólogo. Con las manos ahora demasiado inestables para ordeñar sus vacas, sobrevive gracias a los huevos que su esposa vende.
En la cercana localidad de Juan de Acosta, Mari Echeverría, de 22 años, relató una existencia casi paralela a la de Julieta, su prima hermana (las dos son también primas hermanas de José y Nohora Esther). El año pasado, mientras Julieta bañaba a su tío, ponía música para animarlo y le daba de comer Ensure con una jeringuilla, Mari hacía lo mismo por su madre, que murió un mes después que Nelson. Mari, al igual que Julieta, cuidaba entonces de un bebé y un niño pequeño al mismo tiempo.
Mari era adolescente cuando empezó la enfermedad de su madre, a los 42 años. Eran los días en que Daza, el médico, todavía venía en su furgoneta con medicamentos y palabras tranquilizadoras. Tanto la madre de Mari como Nelson vivieron el resto de sus enfermedades en gran medida sin los medicamentos habituales, como la tetrabenazina para controlar los movimientos y los fármacos psiquiátricos para calmar las emociones y favorecer el sueño. La madre de Mari, una mujer amable y curiosa antes de su enfermedad, a veces lograba hacerse de un cuchillo y lo lanzaba.
Cuando la madre de Mari tuvo fiebre, en sus últimas semanas de vida, Mari la llevó a un hospital de Barranquilla, donde los médicos no reconocieron sus síntomas ni habían oído hablar de su enfermedad. “Me tocó entrar allá y yo escribirles en el computador”, recuerda. Le preguntaron: “‘¿Pero qué tratamiento le ponen a tu mamá? Y yo le dije: ‘¿Yo qué voy a saber qué tratamiento?’”.
A diferencia de sus parientes mayores, que recuerdan una época en la que la enfermedad se ocultaba, se negaba o se atribuía a otras causas, Julieta y Mari crecieron conscientes de que cada una tiene un 50 por ciento de probabilidades de desarrollar Huntington a mediana edad. Ambas optaron por tener hijos, con parejas que comprenden ese riesgo.
Julieta ve el espectro del Huntington con cierta resignación. “Hay peores formas de morir”, dice. Pero tanto ella como Mari participan en el estudio de la Universidad Simón Bolívar. También participan en eventos patrocinados por Factor H, una organización benéfica enfocada en Huntington con actividad en Colombia y Venezuela que se esfuerza por conectar a los investigadores con las familias.
“Queremos perder esa falta de confianza que existe entre las comunidades vulnerables y los médicos”, afirma Ignacio Muñoz-Sanjuán, fundador de Factor H y director ejecutivo de Cajal Neuroscience, una empresa de descubrimiento de fármacos con sede en Seattle. “Y que los médicos e investigadores comprendan que hay seres humanos detrás de lo que intentan hacer”.
Una trampa de atención a la salud
La Fundación CHDI de Princeton, Nueva Jersey, un grupo sin fines de lucro que apoya la investigación de fármacos contra el Huntington en todo el mundo, tiene cada vez más actividad en América Latina. Una de las razones es la genética. “Entre la población europea hemos encontrado una serie de los llamados modificadores, que son objetivos para nuevos fármacos”, explica Cristina Sampaio, directora médica de CHDI. En Latinoamérica, añadió, “podríamos encontrar otros genes que influyan en la evolución de la enfermedad. Estos modificadores podrían convertirse en objetivos”.
Otra razón es la abundancia de personas jóvenes y todavía asintomáticas. CHDI dirige Enroll-HD, un estudio observacional global de personas con enfermedad de Huntington. Aunque la mayoría de sus 25.000 participantes hasta ahora han tenido la enfermedad en estado avanzado, “tenemos una gran necesidad científica de estudiar a personas que estén muy al principio del proceso de la enfermedad”, dijo Sampaio. “América Latina puede ser un lugar donde podamos encontrar más de ellas, y estas son las personas que tienen la mejor oportunidad de beneficiarse del tratamiento”.
Enroll-HD no puede operar en Venezuela, debido a los obstáculos políticos, una infraestructura de atención a la salud degradada y la pobreza extrema. Incluso en Colombia, donde gran parte de la población recibe atención a la salud de calidad, persisten enormes disparidades. Los investigadores de la Universidad Simón Bolívar están deseosos de participar en Enroll-HD, pero queda por determinar quién puede ser estudiado en el marco de la plataforma.
“En estos pueblos, las carreteras son malas y no hay médicos disponibles”, dijo Sampaio, que visitó la zona el año pasado. “No podemos hacer estudios en poblaciones que no tienen acceso a unos cuidados mínimos. Así que podría plantear un dilema estilo Trampa 22. Involucrarlos en la investigación plantea problemas éticos, pero si fueran inscritos, su acceso a la atención mejoraría”. Por ahora, dijo Sampaio, Enroll-HD planea trabajar solo con personas que viven en Barranquilla o que tienen los medios para trasladarse allí.
Muñoz-Sanjuán, médico de Factor H, se mostró optimista en cuanto a que, con esfuerzo, las condiciones podrían mejorar lo suficiente como para permitir que personas como Mari y Julieta se unan algún día a un ensayo clínico.
“El obstáculo en la costa han sido los esfuerzos fragmentados sin un enfoque integral”, dijo. “La gente quiere ayuda desde una perspectiva clínica y social; necesitamos acceso a las personas para desarrollar mejores terapias. Estas dos cosas tienen que estar conectadas”.
Maripaz
Gepsy Ariza, de 34 años, no sabe qué parentesco tiene su familia con otras de la región, solo que son una familia con Huntington. Ella, una joven melancólica, también relata la experiencia de cuidar a una madre enferma, de Daza y sus medicinas, y de haber donado su sangre a unos estudiantes de neurología, para que luego le dijeran que las muestras se habían perdido.
En la polvorienta ciudad costera donde vive, a poca distancia de Barranquilla pero a un universo de distancia de sus comodidades, Ariza ayuda a cuidar de su hermana, Maripaz Ariza. Con solo 30 años, Maripaz tiene unas manos que se abren en abanico y se retuercen como las de una bailarina balinesa, una voz vacilante y un andar rígido y torpe.
Los síntomas de Maripaz aparecieron hace siete años, cuando estaba embarazada de su hija. La enfermedad se manifestó primero como una crisis emocional, recuerda Gepsy. Cuando Maripaz estaba en el hospital esperando para dar a luz, salió corriendo presa del pánico, molesta porque su bebé iba a ser una niña y no el niño que ella esperaba. El personal llamó a los asistentes sociales para que investigaran antes de permitirle ver a su recién nacida.
Los antecedentes familiares con la enfermedad de Huntington nunca se investigaron. A Maripaz le diagnosticaron psicosis. Su comportamiento se ha vuelto más infantil a medida que han progresado sus síntomas motores, lo que la hace parecer más una amiga que una madre para su niñita. Cuando Maripaz recién enfermó, su tío acogió a Maripaz y a su hija en su casa. Más tarde murió de Huntington, y ahora su viuda cuida de ambas.
Poco se sabe de la enfermedad de Huntington y el embarazo, tan raros son los casos en la literatura científica. Siete años después de su enfermedad, Maripaz aún no ha ido a un neurólogo.